I assume we're all interested in similar things.
When we've got a genome with lots of coding sequences in it, we're curious. We want to find out what the most similar known sequences to our nice new coding sequences are, because that might give us some insight into their function. So we can use BLAST for that: just make a conceptual translation of the genecall, a quick command-line and there's your best BLAST match, fresh from the nr database (or wherever). Some skill and judgement later, and you can decide whether you believe it.
But what if that's not enough? What if you've got a few thousand sequences you want to compare? Skill and judgement take time. Or, what if you want to know whether there's an equivalent sequence in another organism? Well, that's where the semantics start to limit your options.
 |
| "Repeating myself" |
Equivalent how?
I find myself asking "What do you mean by equivalent?" quite often, when people ask me to find equivalent sequences for them. Most of the time, they've not thought about it. I know this because they ask me for "sequences with a lot of homology", as though homology comes in variable amounts (it doesn't).After a few minutes, their thoughts tend to clarify towards wanting sequences that are functionally equivalent. They want to know if there's a sequence in another organism that performs the same biochemical function (maybe a chemical interconversion, maybe a binding event) in the same biological context, in an analogous tissue, in response to analogous stimuli.
That's when I usually have to disappoint them, because bioinformatics can't really tell you that. It can give you a candidate, which we can hypothesise has all those properties. But it's still a candidate that you then have to go and demonstrate - experimentally - has those properties.
Sorry about that. No Magic 8-Ball.
One of the quicker, and more reliable ways to identify these candidates in bulk is by reciprocal best BLAST hits (RBBH).
The reciprocal best BLAST hit
RBBH are straightforward to calculate, and require only a slight extension beyond a plain old vanilla one-way BLAST hit. Given the gene complement of genomes A and B, you BLAST the gene products from genome A against those from genome B; and you BLAST the gene products from genome B against those from genome A. Then, you look at your results: the reciprocal best BLAST hits are those where, for sequence SA in genome A and sequence SB in genome B, the best match to SA is SB, and the best match to SB is SA.That's (almost: there can be some technicalities around calling ties or near-ties) all there is to it, but it's best done using a script.
RBBHs perform two very useful functions
- They filter out poor 'best' BLAST matches
- They provide (when comparing the full gene complements of two organisms) a list of candidate orthologous sequences
RBBHs filter out poor BLAST matches

The plot above is a histogram of a few million best one-way BLASTP matches with default settings. They're pairwise searches (genomes 1v2, 1v3, 1v4, ..., 1vn, 2v1, 2v3, ..., 2vn,..., nv(n-1)) between the predicted protein complements of around thirty bacteria from the same genus. It's a plot of density (essentially frequency) against a log of the match bit score, so the horizontal x-axis represents the quality of the match. Bit score is improved by longer alignments with more physico-chemically similar amino acids. The plot is for about 3.5m BLASTP results.
You'll notice a couple of things right away.
Firstly, the distribution is bimodal: there's a large broad peak of matches with decent bit score. These are probably decent matches between pretty similar sequences. But there's a narrow, and noticeable, peak of matches with poor bit score. These are 'best matches' that are short, quite sequence-dissimilar, or both. I think I'd be inclined not to believe them, if I looked closely. We could get rid of them by using a hard threshold of bit score greater than 4 for a believable hit, but that's ugly and arbitrary, and I prefer to avoid that.
We might expect that most of the poor BLASTP matches here result from BLAST just doing its job and finding the best match it can, regardless. That's what it does. It's a search tool and, though it doesn't really want anything, it really wants to find you something that looks like your query. It's a bit like a hunting dog with a creative approach that involves retrieving random objects.
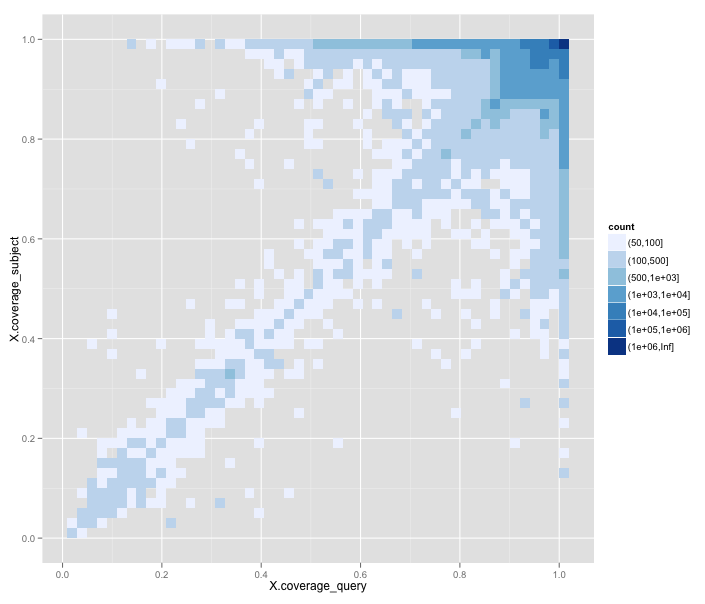
One systematic error we find in this data is obvious when we plot the frequency of 'best matches' on a graph of the percentage alignment coverage of the subject sequence against the coverage of the query sequence:

Here dark blue indicates a very high frequency (it's a log scale), trending down to white for very low frequency of matches. What you can see is that most of the best matches have almost 100% coverage, and sit in the upper right-hand corner. But there's also a diagonal indicating that alignments with n% coverage of the subject sequence tend to have around n% coverage of the query too. But there's a big issue with the lower right triangle, under the diagonal.
Matches in the lower right triangle have alignments that match more of the query sequence than the subject sequence. That is, they represent matches like those of a short protein to a much larger protein. In other words, poor candidates for functional equivalence.
A similar plot of percentage alignment identity against log of bit score shows two clear populations - the same ones as in the histogram above.

The low bit score (poorer) matches are clearly associated with low alignment identity. The larger bit score (better) matches are predominantly clustered near 100% alignment identity. Those low bit score matches - and quite a few moderate bit score matches - have pretty low sequence identity. It looks like we were right not to just use a hard cutoff at bit score = 4; we'd have kept in a bunch of dodgy-looking matches.
So what happens when we look only at the reciprocal best BLAST hits?
Most importantly, we lose the peak of low bit score hits:

That's a sign that those matches were not reliable guides to the equivalence of those sequences. We see that these low-quality matches made up almost all of that lower right triangle of short query matches to long subject sequences:
Even better, we've lost that population of low-identity alignments:

In short, if you use reciprocal best BLAST hits as a filter, you can discard a large number of low-quality alignments and improve your confidence in having found matches to equivalent (in some way) sequences.
RBBHs provide a list of candidate orthologues
This is a straightforward logical argument. I've had it a lot, and proving that it's correct doesn't always seem to convince people.
- Orthologous sequences are sequences that diverged due to speciation. (That is all they are, and orthology does not imply functional equivalence.)
- If the sequences in two organisms diverged only due to speciation, then under normal circumstances the most similar match to one sequence in another species is expected to be its orthologue.
- This is true for both organisms in a pairwise comparison, so each orthologous sequence is the best match to the other: they constitute a reciprocal best match.
However, this argument only works one way. We expect orthologous sequences to be RBBH, but that does not imply that all RBBH are orthologues. I'm not the only person to have noticed this, but it is pretty obvious.
Demonstrating orthology can be quite tricky, and what RBBH gives us is, at best, a set of candidate orthologues that is likely to be larger than the actual set of orthologues.
RBBH do quite well, though. At least two papers:
- Altenhoff, A. M., and C. Dessimoz. 2009. Phylogenetic and functional assessment of orthologs inference projects and methods. PLoS Comp Biol 5:e1000262. doi:10.1371/journal.pcbi.1000262
- Salichos, L., and A. Rokas. 2011. Evaluating Ortholog Prediction Algorithms in a Yeast Model Clade. PLoS ONE 6:e18755. doi:10.1371/journal.pone.0018755
So there we go: reciprocal best BLAST hits - straightforward and useful.
No comments:
Post a Comment